Red Line Walkthrough - Annotating a Genomic Sequence
Annotation adds features and information to a DNA sequence – such as genes and their locations, structures, and functions. DNA Subway’s annotation workflow is divided into four sections:
Create Project:
- Log-in to DNA Subway - unregistered users may 'Enter as Guest'
- Click ‘Annotate a genomic sequence.’ (Red Square); select the 'Web Apollo' version
- For 'Select Organism type' choose 'Plant' and then 'Dicotyledon.'
- from 'Select a sample sequence' chose 'Arabidopsis thaliana (mouse-ear cress) chr1, 75.00 kb'.
- Provide your project with a title, then Click ‘Continue.’
Generate Evidence:
At several stops on the Red Line you will generate mathematical evidence (using the repeat and gene prediction algorithms) and biological evidence (derived from BLAST homology searches).
Find and mask repeats
- Click ‘RepeatMasker’. Wait until the flashing icon displays ‘V.’ (view)
- Click ‘RepeatMasker’ again to view the results.
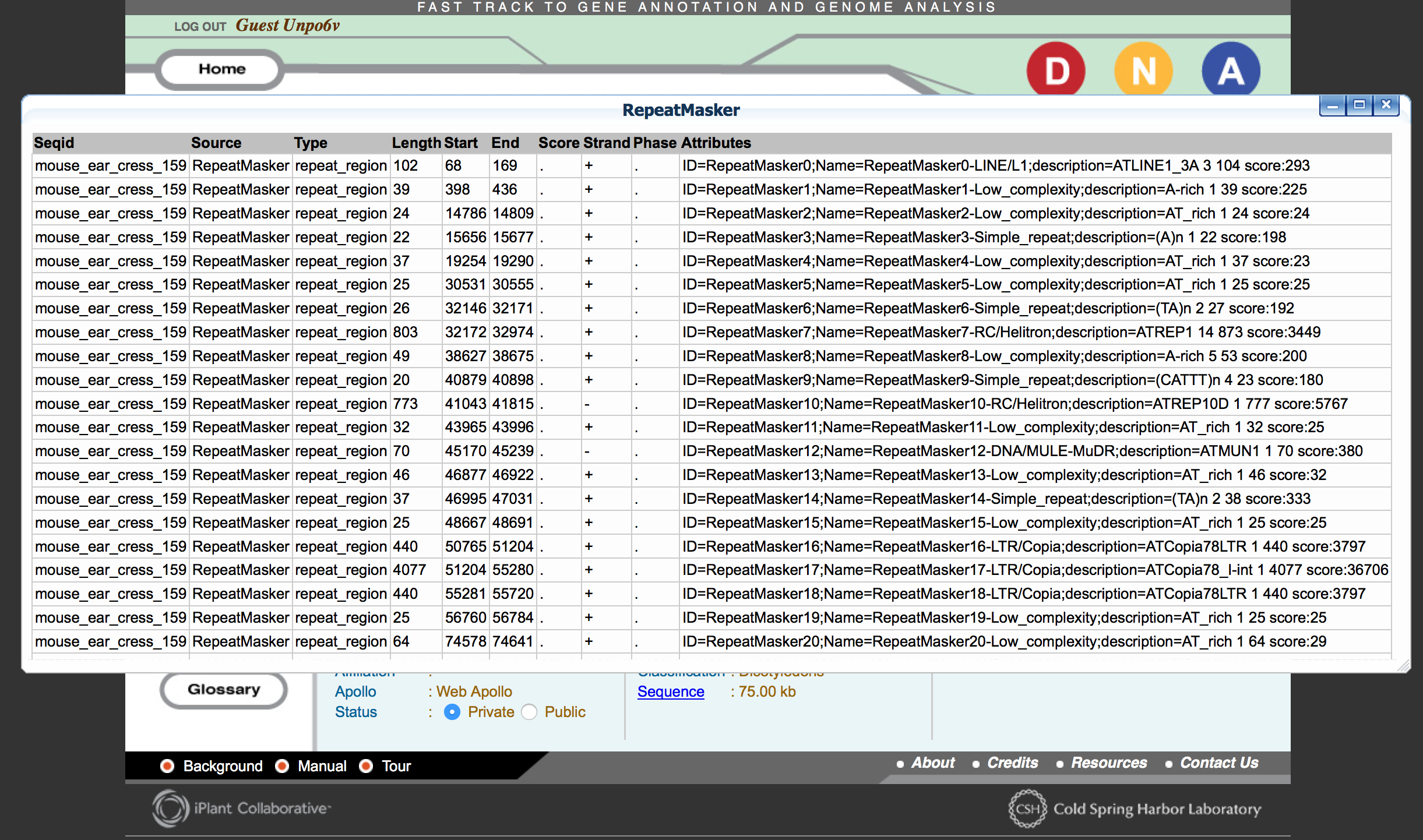
Questions:
- How many hits were detected in your sample?
- RepeatMasker reports the length of the repetitive sequences
(Length) as well as the class (Attributes).
- What is the average length of sequences identified as “simple repeats”?
- What is the average length of sequences identified as “low complexity”?
- What is the total percentage of repetitive DNA in your sequence? (Sum of the length of all repetitive sequence / sequence length (75 kb)
Predict genes (de novo)
- Click ‘Augustus’ and wait until a green ‘V’ icon appears.
- Click ‘Augustus’ again to view a table of results.
- Repeat (one-at-a-time) steps 1-3 with ‘FGenesH’, ‘Snap.’
Questions:
-
Look at the ‘Type’ column in the gene prediction report. Find the gene labeled Name=AUGUSTUS012;ID=g12 and copy down the gene’s ‘start’ (i.e. the starting basepair). How many exons are predicted? give their starting locations.
Gene Predictor Start Exon AUGUSTUS 267 1 AUGUSTUS 323 2 AUGUSTUS AUGUSTUS AUGUSTUS AUGUSTUS AUGUSTUS AUGUSTUS AUGUSTUS AUGUSTUS -
Based on the chart in question 4, did all the gene predictors yield genes starting at the same location? Did all the gene predictions have the same number of exons?
- Looking at the number of results returned by tRNA Scan, why are they so different from results made by other predictors? Are their places in the genome where tRNAs are more or less densely concentrated?
Visualize predicted genes in a Genome Browser
- Click ‘JBrowse’ and allow browser to load.
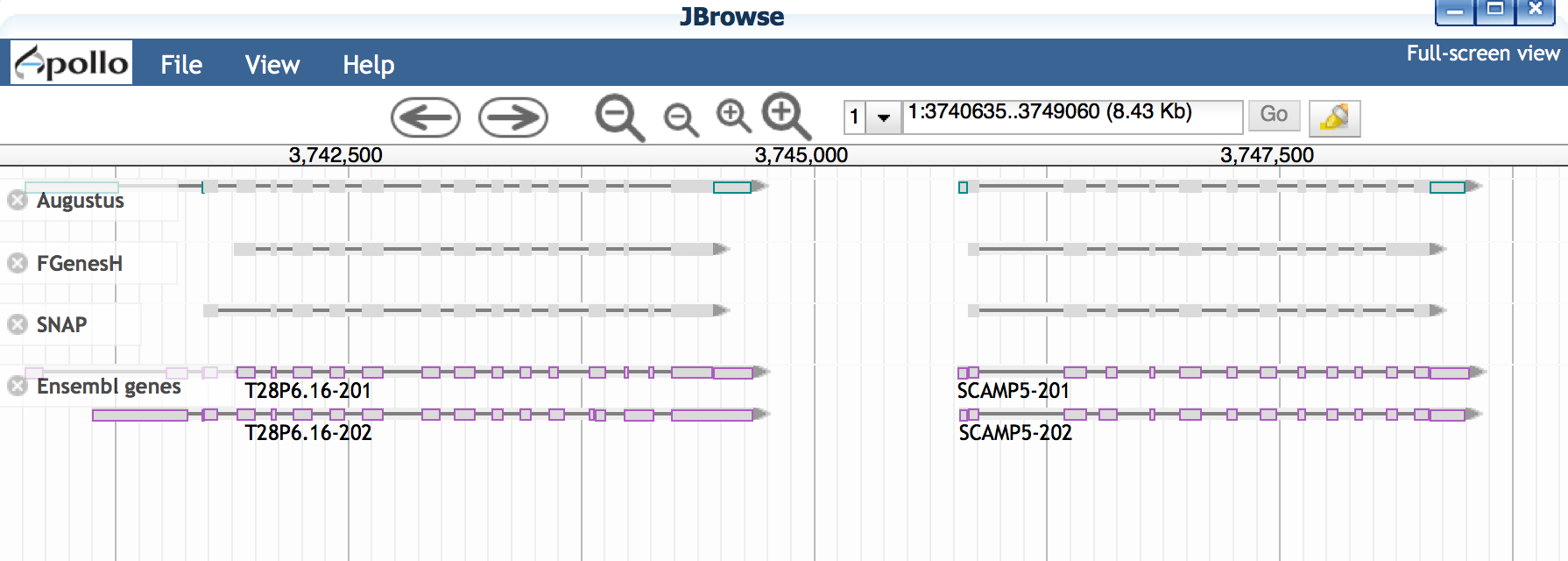
- Zoom into a region (for example, paste the region 1:3740638..3749063 into the location window.
- Examine gene details by double-clicking on a gene to select; then right-click to open the 'View Details' menu.
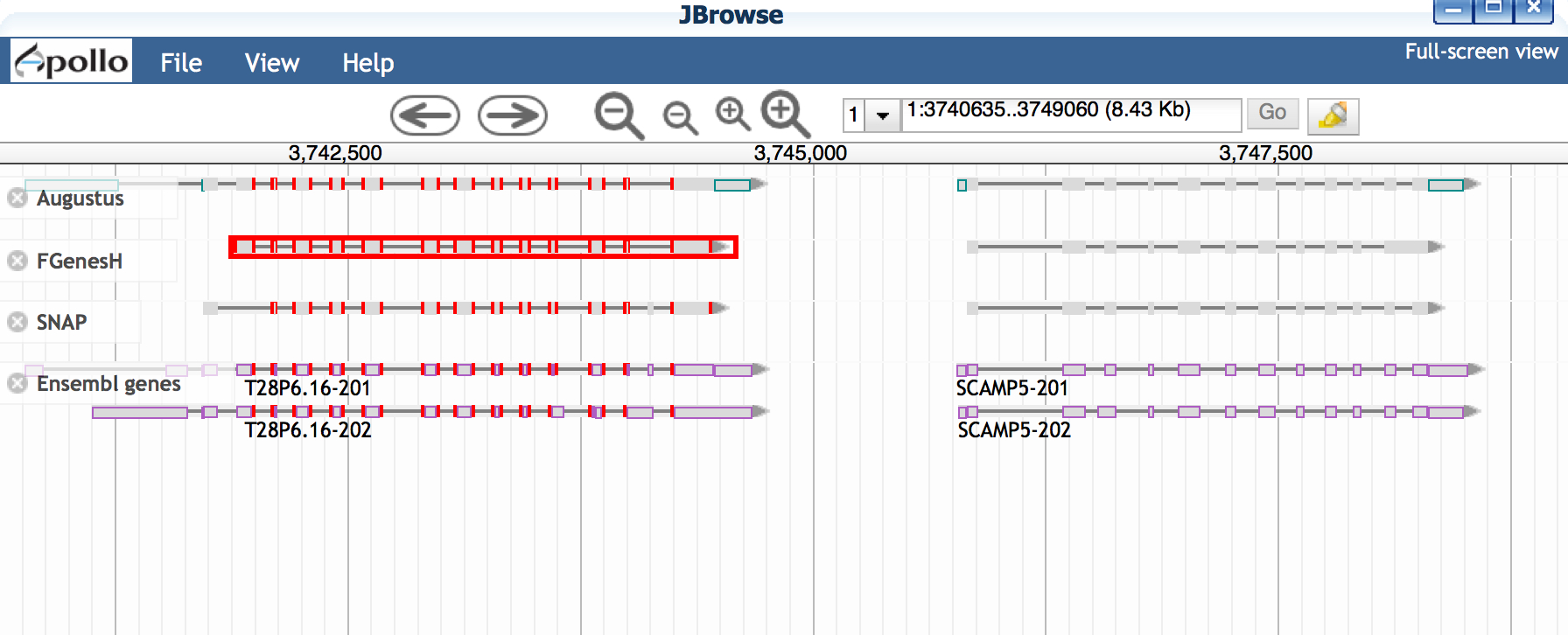
Questions:
- Do all the gene predictors agree with each other?
- Which gene predictions seem to match the Ensemble genes most closely?
Search databases
DNA Subway searches customized versions of UniGene and UniProt that contain only validated plant proteins, and are free of predicted or hypothetical proteins.
- Click ‘BLASTN’; wait until the flashing icon displays ‘V’ (view)
- Click ‘BLASTN’ again to view the results.
- Click ‘BLASTX’; wait until the flashing icon displays ‘V’ (view)
- Click ‘BLASTX’ again to view the results.
- Click on 'JBrowse' and then click 'Full-screen View' in the upper-left
- In the 'Available Tracks' menu, add the Blastn and Blastx tracks.
Questions
- Both BLASTN and BLASTX returns the ‘Length’ of your resulting matches. Do you notice differences in the average lengths of BLASTN and BLASTX matches? Explain.
- Under ‘Type’ both BLASTN and BLASTX returns ‘match’ and ‘match_part.’ ‘Match’ is describing the overall length of a single match, but individual significant matches may be fragmented, i.e. ‘match_part.’ Do BLASTN and BLASTX return ‘match’ and ‘match_part’ results in different frequencies? Explain.
Build gene models (WebApollo)
Apollo is an extension of JBrowse which allows the user to build and edit gene models. Apollo has a number of features but in this tutorial, we will give brief intro covering the conceptual steps.
Import Blastn model to match for transcript length
Blast searches are matched against UniGene(blastn) and UniProt(blasts). UniGen models are derived from cDNA and ESTs (transcriptome evidence) produced by experiment. (http://www.ncbi.nlm.nih.gov/UniGene/help.cgi?item=build2)
- Open Apollo and zoom into a region of interest (e.g. 1:3793981..3802033)
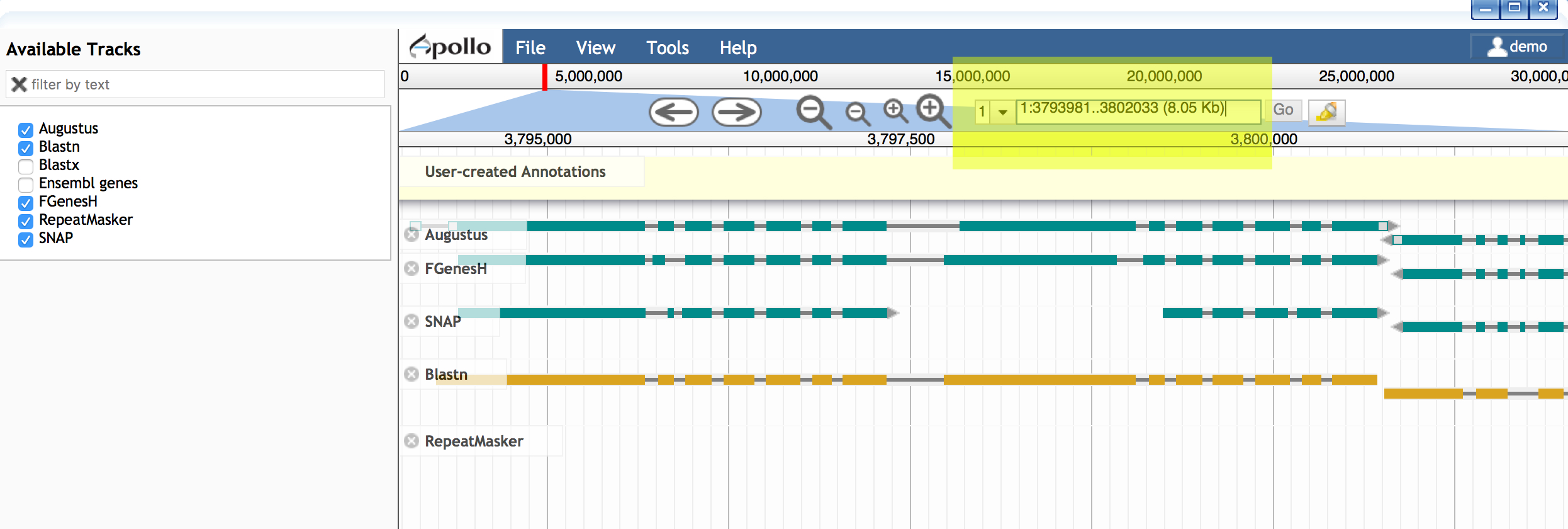
- Ensure at least the following tracks are selected (on):
- Augustus (and other gene predictors: FGenesH, SNAP, etc.)
- Blastn
- Drag the Blastn result into the yellow 'User-created Annotations'section.
Select a scaffold model
Use transcriptome evidence (UniGene - BLASTN) to select the best possible gene model for a scaffold. If no gene model exists or significantly reflects the UniGene model, use the UniGene model itself as a scaffold
- Drag a plausible model into the yellow 'User-created Annotations' - in this case we will choose the Augustus model; double-click the Augustus model to select the entire model and drag into 'User-created Annotations'.
- Adjust the Augustus model to match the 5' and 3' configuration of the blastn model
- Delete the extraneous 5' exon (single-click to select; right-click to delete)
- Adjust the new 5' end to match the length of the blastn-derrived transcript
- Adjust the 3' end of the Augustus-derrived model (single-click to select; use mouse to adjust the model length)
Edit model for splice sites and variants
Protein and EST data can be used to examine possible alternative transcripts. Proteins give clues to the actual length of the translated protein at that locus and its reading frame. Like full length cDNAs, ESTs give valuable information on transcript diversity. ESTs are generated by high throughput methods, and although the data may be fragmentary, it may capture biologically relevant information about splice variants.
- Turn on the blastx track
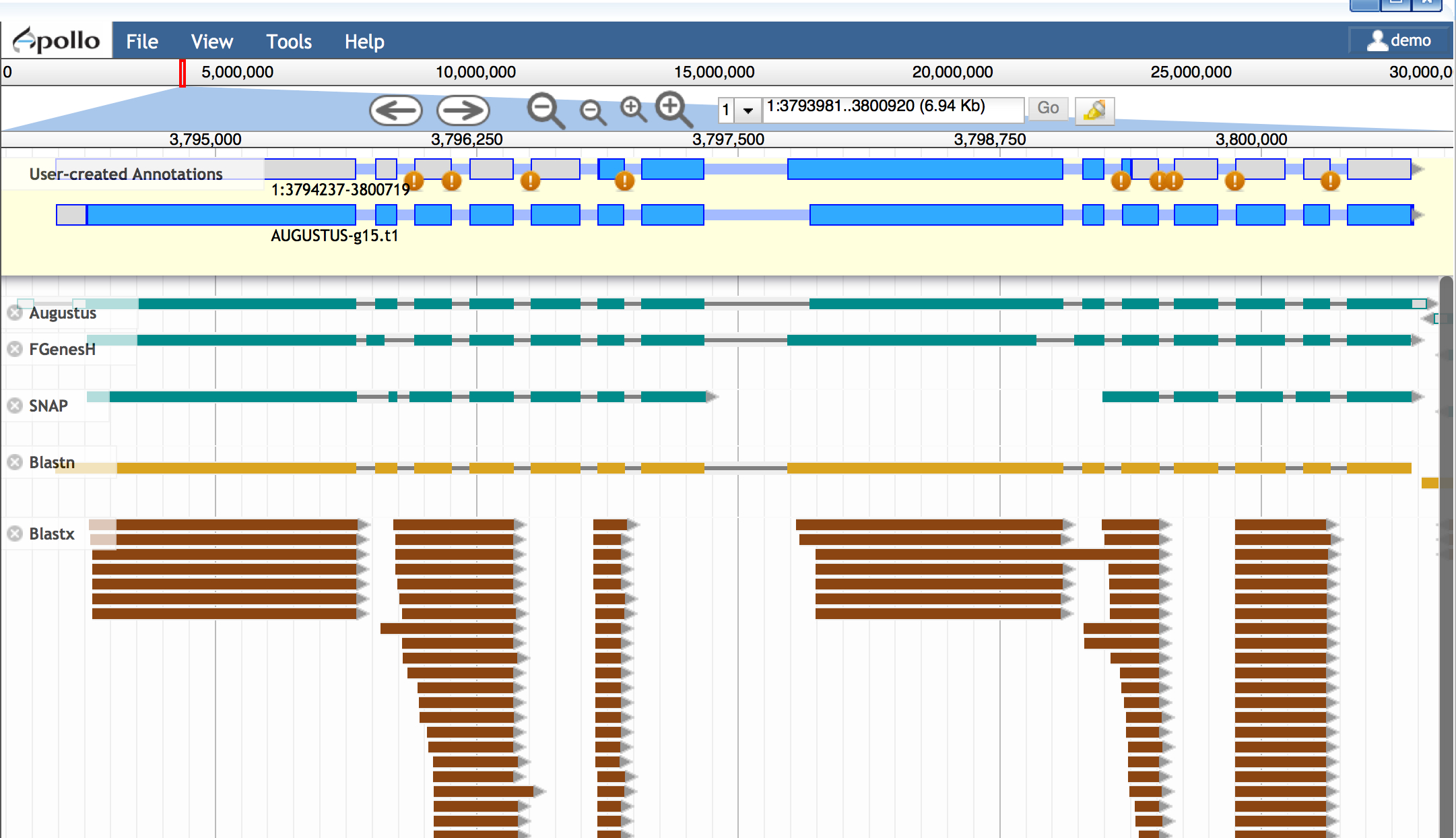
- Examine the additional evidence to consider making adjustments to your Augustus-derrived model. If you wish to make additional isoforms of your gene:
- Double-click to select the entire Augustus-derrived model
- Right-click on the model to duplicate
- Make adjustments to the model as desired
Determine translation start/stop sites
After making your adjustments, you can confirm that your gene model(s) represents the longest possible transcripts:
- Double-click the model; right-click and select 'Set longest ORF'
Compare gene model(s) with existing annotations
After making your gene models you can compare them with existing annotations by turning on the 'Ensemble genes' track.
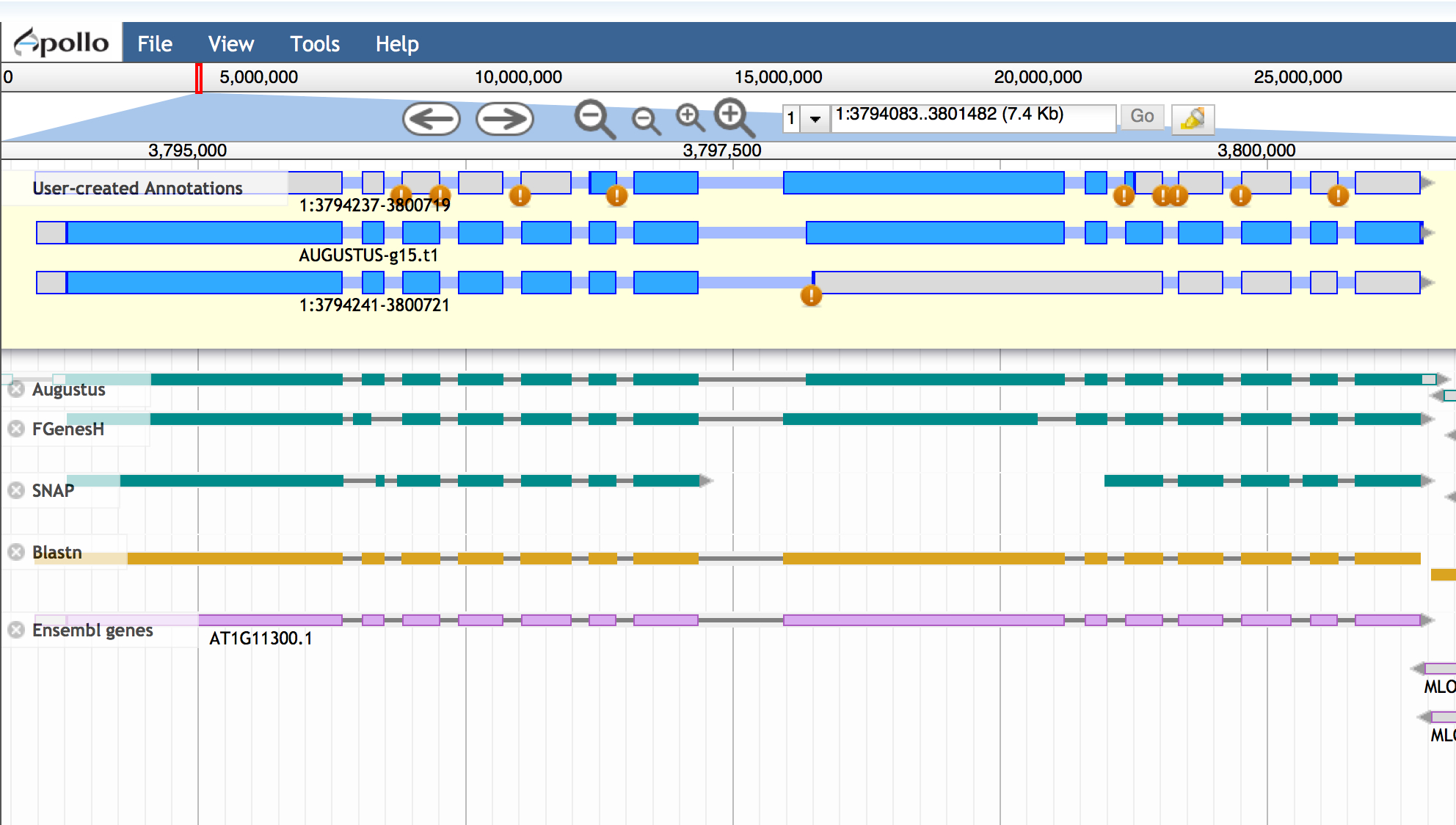
In this case, our work confirms the first gene model made, but a potential isoform supported by blastx data is likely incorrect.